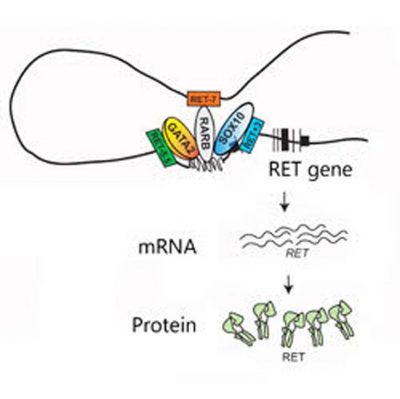
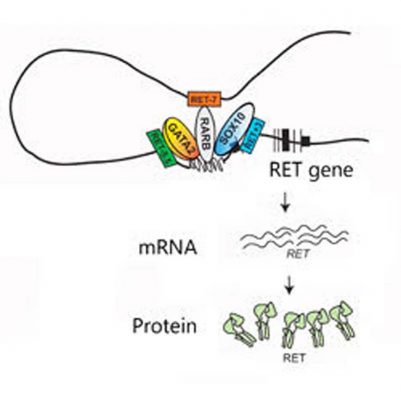
Mutations in RET’s three enhancers (RET -7, RET -5.5 and RET +3) can disrupt the binding of their transcription factors (RARB, GATA2 and SOX10, respectively) and alter the activation of the RET gene and the production of RET mRNA and protein. (Credit: Cell Press)
Finding has implications for other diseases involving multiple genes
By Catherine Gara
Baltimore, MD, Sept. 29, 2016 – Scientists at Johns Hopkins Medicine say they are one step closer to understanding the genetic mechanism of a rare, complex, multiple-gene disorder called Hirschsprung’s disease.
The results of their latest study suggest that many patients develop the disease when multiple mutations in gene regulatory sequences of a specific gene combine to destroy the normal cooperative function of a whole network of genes.
The investigators say they expect that similarly dysfunctional gene networks influence the onset and progress of other diseases that involve multiple genes, and that dissecting these networks may eventually lead to improved diagnosis and treatment options for patients with disorders like cancer, diabetes and high blood pressure.
A summary of the study was published online on Sept. 29 in the journal Cell.
“We tend to think of genes as operating on their own, but they can’t and don’t,” says Aravinda Chakravarti, PhD, who led the study and has pioneered research on the genetics of so-called multigenic and complex diseases.
“Instead,” he says, “they exist within ‘families’ of genes and regulatory elements that live in the same section of the chromosome and interact frequently, each having its own job but all working together toward one goal: making a gene function. If you perturb one individual, everyone else is affected.”
Chakravarti, a professor in the Johns Hopkins University School of Medicine’s McKusick-Nathans Institute of Genetic Medicine, has studied Hirschsprung’s disease since 1990 because it is a relatively easy-to-investigate model for disorders with complex genetic causes.
For example, the disorder has a wide range of severities and symptoms but a single affected organ: the colon. Overall, the disease occurs when nerve cells fail to connect to the muscles of the colon during fetal development, leaving a child unable to pass stool.
It affects about one in 5,000 newborns and can usually be resolved surgically, but 30 to 50 percent of patients still have complications throughout their lives.
In 2002 and 2015, Chakravarti’s team performed genomewide association studies (GWAS) to identify common genetic variants associated with the disease. Further research showed that variations in the RET gene and its regulatory elements were standouts among patients.
The researchers found then that almost every person with Hirschsprung’s has at least one RET-associated variant. However, because many healthy people without Hirschsprung’s do too, the causal mechanism remained a mystery and led them to look for multifactorial causes.
They decided to focus on eight single “letter” changes in the genetic code, or SNPs, known to occur in specific regions near the RET gene. These so-called gene enhancers act like dimmer switches, turning up or down the activity, or transcription, of nearby genes by binding and properly positioning proteins called transcription factors.
After swapping out the RET gene for a reporter gene, the scientists ran a series of tests in a human nerve cell line and showed that only three of the SNPs, each in a different enhancer, were capable of decreasing gene activity on their own by weakening the binding of that enhancer’s transcription factor.
When the scientists tested the three SNPs in different combinations, they found that although none of the enhancers are known to interact with each other directly, a general trend appeared: The more enhancers were affected, the lower the reporter gene activity. When the team looked at the frequency of these regulatory variants among 346 patients with Hirschsprung’s disease and 732 healthy controls, the trend was confirmed. Those with all three SNPs had a fourfold higher risk of having the disease.
In further experiments with mice, the researchers say they found that the three enhancers drive gene activity at different stages in the developing fetal gut and were able to identify which transcription factors bind to them. Then, they deleted the RET gene from mice and examined how its “gene family” responded.
The genes for two of the transcription factors that bind its enhancers — and for another protein that breaks down RET to stop its signaling — were less active when RET was missing. But genes for a protein that works with RET on the cell’s surface and the signaling molecules that bind to RET were increased in activity. Data from human cells also showed that decreasing the levels of the transcription factors decreased the levels of RET.
“These findings help explain why RET-associated variants are necessary but not sufficient to cause Hirschsprung’s disease,” says Sumantra Chatterjee, PhD, the first author of the newly published research paper. “It provides a way forward for studying genetic synergy in the context of health disorders.”
Chakravarti adds, “We’ve been able to show that these genes are intimately related to each other not because of similarities in sequence or function or even location in our genome, but because they all work together to execute one function: getting cells to become nerve cells for the colon.”
Other authors of the report include Ashish Kapoor, Dallas Auer, Dongwon Lee and Courtney Berrios of the Johns Hopkins University School of Medicine; Jennifer Akiyama and Len Pennacchio of the Lawrence Berkeley National Laboratory; and Stacey Gabriel of the Broad Institute.
This work was supported by grants from the National Institutes of Health, the National Institute of Dental and Craniofacial Research, National Human Genome Research Institute and the Department of Energy.





